Ruling the Future: How the ACLA’s LDT Court Victory and Europe’s IVDR Are Shaping Bioanalytical Laboratories
by Chad Briscoe, Executive Vice President, Global Bioanalytical Services, Celerion
Someone whom I’ve worked with in the past and have had an opportunity to mentor in the bioanalytical field for some time recently asked me about the U.S. federal court ruling on LDTs. I realized I don’t really understand it as well as I would like to or as well as I should, and that I hadn’t been as “up-to-speed” on the current status of LDTs as used in the US or Europe. I decided to dig in and pull together a blog to help me learn and also help provide a guide for others that may want something that summarizes the current situation as I understand it.
In a landmark legal decision announced by the ACLA (American Clinical Laboratory Association) on March 31, a U.S. federal court vacated the FDA’s attempt to regulate Laboratory Developed Tests (LDTs) as medical devices. This preserved the long-standing CLIA oversight. Essentially preserving the status quo.
Meanwhile, across the Atlantic, the European Union’s In Vitro Diagnostic Regulation (IVDR) has been tightening control over diagnostic innovation. These two contrasting regulatory directions are reshaping the landscape for clinical and bioanalytical laboratories. Here’s my interpretation of what it means, why it matters, and how labs (in particular bioanalytical labs) can stay ahead.
Laboratory-Developed Tests (LDTs) have become foundational to the rapid evolution of diagnostic science. In recent years, due to the increasing complexity of LDTs and the focus on biomarkers in traditional bioanalytical labs, this has become important to laboratories beyond just the traditional Clinical Laboratories, central laboratories, and local hospital labs. These custom assays—designed, manufactured, and used within a single or small number of laboratories—often address unmet clinical needs, are used to support clinical trials and new treatment options. These have become key tools in affordable precision medicine. However, their regulation has been in flux for over a decade, driven largely by activities in the United States and European Union.
In the U.S., a federal court recently vacated the FDA’s 2024 final rule that sought to classify LDTs as medical devices. This decision, brought forward by the American Clinical Laboratory Association (ACLA), reasserts CLIA (Clinical Laboratory Improvement Amendments) as the primary regulatory authority over LDTs. In Europe, however, the new In Vitro Diagnostic Regulation (IVDR) has taken the opposite approach, intensifying regulatory oversight and requiring an onerous approval process with extensive documentation, validation, and risk classification for all diagnostic tests—including in-house laboratory tests.
Under the previous regulation, about 20% of laboratory tests were required to follow this path, but this has now increased to about 80% under IVDR.
Implications of FDA Regulating LDTs (If ACLA lawsuit had failed)

The FDA’s now-overturned rule would have subjected LDTs to the same premarket review, labeling, and post-market surveillance required for commercial in vitro diagnostic devices, developed for a mass market. Critics, including ACLA and the Association for Molecular Pathology (AMP), argued that this overstepped statutory bounds, stifled innovation, and jeopardized access to critical diagnostics.
The court’s ruling provides:
- Regulatory Stability: Labs may continue operating under CLIA without additional FDA burdens.
- Innovation Breathing Room: LDTs can still be developed and deployed quickly, often in response to urgent or rare clinical needs.
- Continued Responsibility: While FDA oversight is paused, labs must continue to maintain rigorous internal validation practices and high-quality standards.
For bioanalytical labs, this ruling helps sustain agile assay development, enabling continued support for clinical trials, specialty testing, patient stratification, patient enrollment, and companion diagnostics.
The EU’s IVDR, which came fully into effect in May 2022, represents a complete overhaul of how diagnostics are regulated. Replacing the former IVDD, the IVDR imposes:
- Risk-Based Classification (Class A to D) – though most LDTs are highly complex and therefore high risk
- Performance Evaluation Requirements – requiring navigating a complex network of national Notified Bodies with varying interpretations of IVDR
- Strict Use Conditions for In-House Tests – Typically, only hospital laboratories qualify when the test is used within the hospital.
- Mandatory Quality Management Systems – ISO15189 is suggested, but accreditation bodies struggle to accredit non-routine clinical laboratories
- Market Justification for Each In-House Test – requirement for regulatory review for each assay by a Notified Body
In stark contrast to the U.S. model, the IVDR now requires labs to prove that no CE-marked equivalent exists before using an LDT. Labs must also implement formal procedures for ongoing safety, performance monitoring, and clinical evidence gathering.
Key Differences at a Glance
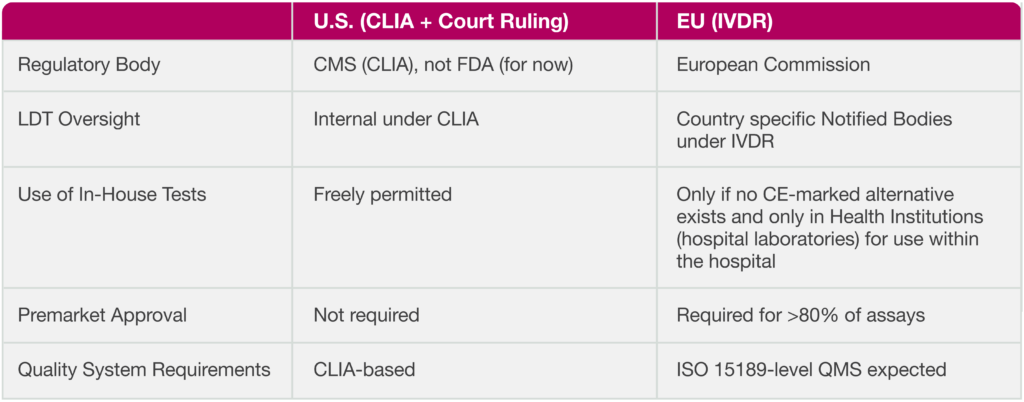
For labs operating in clinical trials, translational science, or patient diagnostics, these changes affect everything from compliance planning to assay design and budgeting.
In the U.S.:
- Labs retain operational flexibility.
- No new device-level regulatory filings required.
- Opportunity for rapid test iteration and clinical trial support.
In the EU:
- Labs face new regulatory hurdles.
- In-house test development is resource-intensive.
- There may be delays or discontinuation of certain novel assays due to compliance costs.
- Patient stratification for clinical trials will be reduced or stopped unless absolutely necessary.
Multinational labs performing LDTs must now manage dual compliance strategies:
- In the U.S., keep internal systems CLIA-compliant while monitoring the policy landscape (e.g., VALID Act proposals).
- In the EU, prepare for full IVDR implementation—especially if supporting EU-based clinical trials or patient testing.
Labs performing cross-border testing (e.g., EU patients using U.S.-based services) will need to harmonize quality, documentation, and reporting standards to remain compliant with IVDR, even if physically located outside the EU. Laboratories outside the EU struggle to find the correct national Notified Body that will review and approve their IVD assay.
The divergence between U.S. and EU regulation poses challenges—but also raises important questions:
- Can future legislation, such as the VALID Act in the U.S., find a middle ground between oversight and innovation?
- Will the EU refine or delay IVDR enforcement based on implementation feedback?
- How can international standards be aligned to ease burdens for global labs?
Industry advocacy groups like ACLA and AMP are already working toward legislative solutions, while European regulators are issuing guidance to clarify IVDR expectations. Collaboration across borders will be key to future success.
Whether you’re a clinical lab director, regulatory affairs lead, or scientist in a bioanalytical CRO, the message is clear: the regulatory environment for diagnostics is evolving—rapidly and asymmetrically.
• In the U.S., the recent court decision protects flexibility and self-regulation—for now.
• In Europe, IVDR brings new rigor and central oversight—but also risks of reduced innovation.
• Globally, labs must adapt or risk falling behind.
As science continues to evolve and offers the opportunity for personalized medicine, regulatory oversight shouldn’t stand in the way of enabling it; instead, it should find a way to accelerate the safe implementation. In our industry, public health regulations that enable advanced clinical trial designs and early patient access to innovative new medicines are just as critical as scientific advancement.
Now is the time for bioanalytical labs to strengthen their quality systems, shake the fear of innovative solutions, and advocate for smart, science-driven oversight that puts patients first.